Distance-based methods
The first step of any distance method consists in constructing a matrix of distances. Launch dnadist (command prompt or double click) and specify yeast1.txt as input file. dnadist's interactive menu looks like given on the right.
The calculation of distances depends on the evolutionary model assumed. Here we want to keep it as simple as possible, and therefore we'd like to run our analyses with the Jukes-Cantor (JC) model, which assumes that all nucleotide changes are equally probable. Type "D" until this option appears, then accept with "Y".

The outfile looks like this:
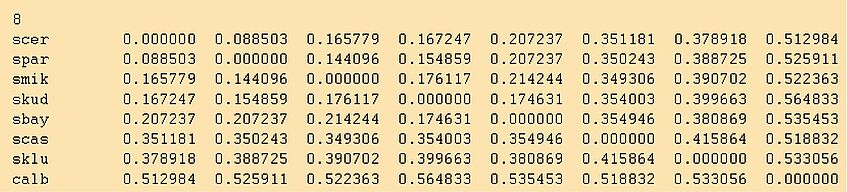
IMPORTANT: Did you rename the outfile? A good new name would be yeast1.dist. Otherwise if you tell neighbor to use outfile as its input file, terrible things will happen. Neighbor first opens outfile as an output file, thus erasing it. When it then tries to read from this empty outfile a psychological crisis ensues!
Now that we have a matrix of pairwise distances, we can run any method for reconstructing a tree that fits well this matrix. For simplicity, we use Neighbor Joining: launch neighbor (command prompt or double click) and specify yeast1.dist as input file.
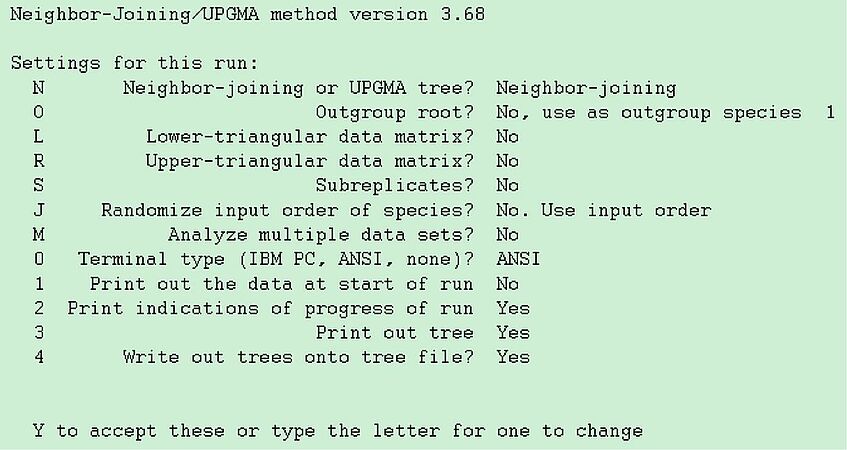
Again the options for neighbor are selected through a menu, which looks like given on the left side.
Notice that option "o" would allow you to specify Candida albicans as an outgroup, but you don't need to do this, as it is the first sequence to appear in yeast1.txt. The other options are mostly uninteresting, so accept by typing "Y" and look at the outfile.
- Q2. Any interesting difference with the tree you previously obtained with dnapars?