Quality assessment of sequence data
Modern sequencing technologies can generate a massive number of sequence reads in a single experiment. However, each instrument will generate different types and amounts of error. Therefore, it is necessary to understand, identify and exclude error-types that may impact downstream analysis.
Our objective here is to understand some relevant properties of raw sequence data. We will focus on properties such as length, quality scores and base distribution in order to assess the quality of the data and discard low quality or uninformative reads.
VBCF provides .bam files, which need to be transformed to .fastq files for example with bam2fastq, but this is not covered here. We start directly from .fastq files.
Fastqc currently is not working in the vetgrid cluster. You can however run it in your local computer. For the purpose of this practical I will just demonstrate myself how it works.
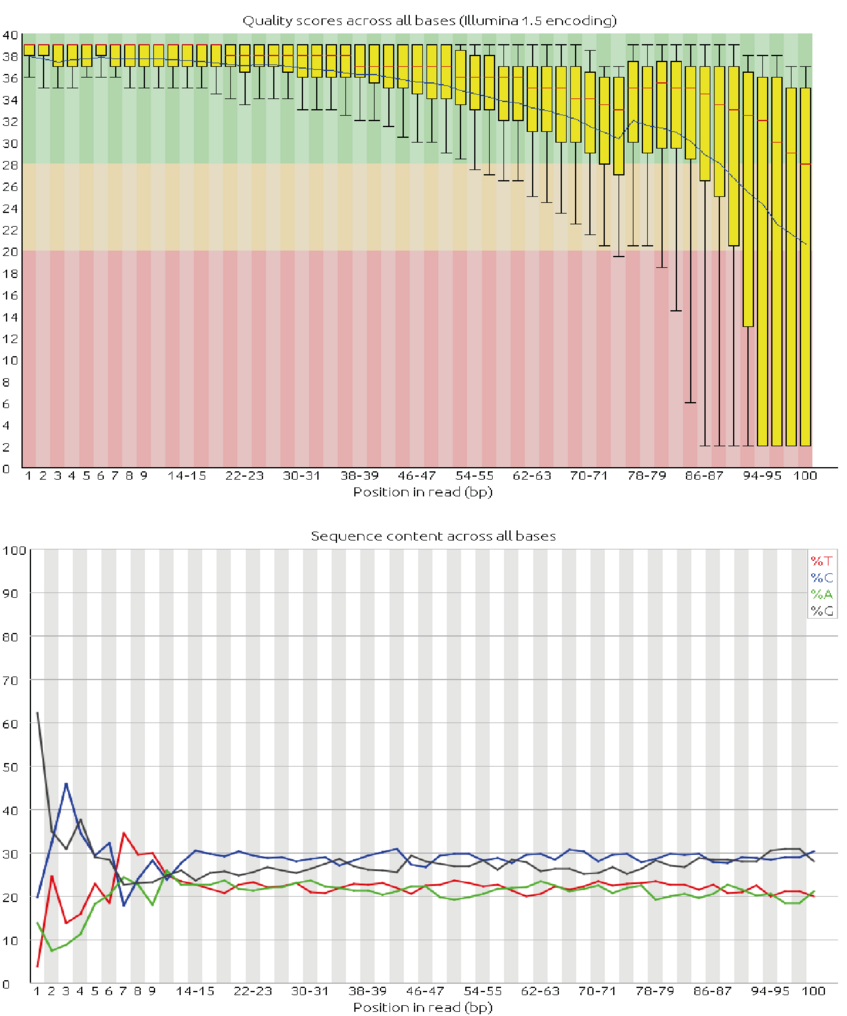
- Have a look at the numbers output on the Basic Statistics page. How many sequences do we have? What is the sequence length? And the GC content?
- Examine the Per base sequence quality and Per sequence quality scores. FastQC points out a problem with a red X and a potential problem with an orange !. Do you think this run gave good quality sequences?
- Examine the Per base sequence content, Per base GC content and Per sequence GC content pages. Do you think we should worry about it in this particular case?
- Examine the Overrepresented sequences page. Why does FastQC give a warning message?
- Let’s have a look also at the file inc_small.2.fq. How do the two files compare?
Quality filtering/trimming
Trimmomatic is a fast, multithreaded command line tool that can be used to trim and filter Illumina (fastq) data as well as to remove adapters. It works with both paired-end and single-end fastq files, and files compressed (e.g., fq.gz) are also supported. The selection of trimming steps and their associated parameters are supplied on the command line.
ILLUMINACLIP: Cut adapter and other illumina-specific sequences from the read.
SLIDINGWINDOW: Performs a sliding window trimming approach. It starts scanning at the 5' end and clips the read once the average quality within the window falls below a threshold.
MAXINFO: An adaptive quality trimmer which balances read length and error rate to maximise the value of each read
LEADING: Cut bases off the start of a read, if below a threshold quality
TRAILING: Cut bases off the end of a read, if below a threshold quality
CROP: Cut the read to a specified length by removing bases from the end
HEADCROP: Cut the specified number of bases from the start of the read
MINLEN:: Drop the read if it is below a specified length
AVGQUAL: Drop the read if the average quality is below the specified level
The results will be 4 files in the folder /YourName/Trimmed/, out of which 2 are paired and 2 are single.
In the vetgrid10 cluster:
cd ~/students/YourName/
java -jar ~/bin/trinityrnaseq-v2.15.1/trinity-plugins/Trimmomatic/trimmomatic.jar PE -phred64 ./inc_small.1.fq ./inc_small.2.fq ./Trimmed/incTrimP.1.fq ./Trimmed/incTrimS.1.fq ./Trimmed/incTrimP.2.fq ./Trimmed/incTrimS.2.fq ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10:2:True LEADING:13 TRAILING:13 SLIDINGWINDOW:4:20 MINLEN:36
- It is recomendable to check the results with FastQC and vary the options to see what the effect is.